
Emergence of resistance to antimalarial drugs has become a major hurdle in the successful treatment of the infection, and has contributed significantly to global malaria-related mortality.[1] Till date, drug resistance has been documented in P. falciparum, P. vivax, and P. malariae.[1] P. falciparum has developed resistance to nearly all antimalarial drugs currently in use; P. vivax has been found to be resistant to chloroquine and primaquine; and P. Malariae has been reported to be resistant to chloroquine and pyrimethamine in some areas.[1-3]
In 1967, WHO defined drug resistance as the ability of the parasite strain to survive or multiply despite the administration and absorption of a drug given in doses equal to or higher than those usually recommended but within the tolerance of the subject. This definition was later modified to include the sentence: “The form of the drug active against the parasite must be able to gain access to the parasite or the infected erythrocyte for the duration of the time necessary for its normal action”. This addition took into account a new development in the understanding of human metabolism of sulfonamide. As the pharmacokinetics of antimalarial medicines varies widely among individuals, the definition of resistance is enhanced if the concentration profile of the active drug concerned is also taken into consideration. For example, in the case of a prodrug, which is not active in the ingested form and requires chemical conversion by metabolic processes to become pharmacologically active, the definition should also include a requirement for a “normal” profile of the biologically active metabolite. Because of the particular mode of action of artemisinins, the definition of resistance may require further discussion and clarification.[4]
Treatment failure is defined as an inability to clear malarial parasitaemia or resolve clinical symptoms despite administration of an antimalarial medicine. Treatment failure is not, however, always due to drug resistance, and many factors can contribute, mainly by reducing drug concentrations. These factors include incorrect dosage, poor patient compliance in respect of either dose or duration of treatment, poor drug quality and drug interactions. Even after supervised administration of a full regimen of an antimalarial medicine, individual variations in pharmacokinetics might also lead to treatment failure because of poor absorption, rapid elimination (e.g. diarrhoea or vomiting) or poor biotransformation of prodrugs.[4]
Multidrug resistance of P. Falciparum is seen when the parasite is resistant to more than two operational antimalarial compounds of different chemical classes and modes of action. Generally, the two classes first affected are the 4-aminoquinolines and the antifolates (diaminopyrimidine, sulfonamides). Drug resistance results in a delay in or failure to clear asexual parasites from the blood, which allows production of the gametocytes that are responsible for transmission of the resistant genotype.[4]
Development of Resistance
The malaria parasite is well known for its frequent, de novo mutations, mostly single, and sometimes multiple. In the presence of heavy infection and inadequate drug levels, the resistant mutations survive and propagate. Development of resistance requires a high grade of parasitemia, coupled with low or inadequate drug levels. Most cases of resistance have emerged out of SE Asia region. This region is known for low transmission and low immunity that lead to high parasitemia; it also has a long history of indiscriminate use of different antimalarial drugs. In such low-transmission areas, most malaria infections are symptomatic, and therefore proportionally more people receive treatment, providing more opportunities for selection of resistant strains. One study also suggested that P. falciparum in South-East Asia has an inherent propensity to develop drug resistance through genetic mutation. Areas with very high transmission, such as Africa, appear to be less susceptible to the emergence of drug resistance. In these areas, infections are acquired repeatedly throughout life, resulting in partial immunity (“premunition”), that in turn controls the infection, usually at levels below those that cause symptoms. Asymptomatic infection and often non-availability of drugs in these areas mean that these patients do not receive antimalarial drugs, and hence the chances of development of resistance are lower.[4] Immunity acts by non-selectively eliminating blood-stage parasites, including the rare de novo resistant mutants, and also improves cure rates, even with failing drugs, thereby reducing the relative transmission advantage of resistant parasites. Furthermore, complex polyclonal infections in semi-immune people allow possible outbreeding of multigenic resistance mechanisms or competition in the host or the mosquito between less-fit resistant strains and more-fit sensitive strains.[4]
 Drug pressure leads to higher gametocyte release, and this facilitates the propagation of the resistant mutants that have escaped the drugs. Failure to use primaquine as a gametocytocidal agent for P. falciparum further aids such spread of resistance. Therefore, gametocyte production from the recrudescent resistant infection must be prevented by administration of early, appropriate treatment [4], combined with primaquine.
Drug pressure leads to higher gametocyte release, and this facilitates the propagation of the resistant mutants that have escaped the drugs. Failure to use primaquine as a gametocytocidal agent for P. falciparum further aids such spread of resistance. Therefore, gametocyte production from the recrudescent resistant infection must be prevented by administration of early, appropriate treatment [4], combined with primaquine.
Administration of drugs with long elimination phases facilitates the spread of resistant mutant malaria parasites. The residual antimalarial activity that is present during the post-treatment period serves as a “selective filter”, which prevents infection by sensitive parasites but allows infection by resistant parasites. Drugs such as chloroquine, mefloquine and piperaquine, which persist in the blood for months, provide a selective filter long after their administration has ceased.[4]
Factors that promote the development of drug resistance are more intense with P. falciparum compared to P. vivax and this explains the higher incidence of resistance in P. falciparum.
Mechanisms of resistance
The biochemical mechanism of resistance has been well understood in cases of chloroquine, the antifolates, and atovaquone. The chloroquine-resistant strains of P. falciparum tend to accumulate the drug less efficiently than the sensitive ones. Polymorphism in the pfcrt (for chloroquine resistance transporter) gene, particularly the one amino acid change, K76T, located in the first transmembrane domain, has been found consistently in chloroquine-resistant P. falciparum parasites. This critical K76T mutation could possibly alter the selectivity of CRT such that chloroquine more efficiently exits the food vacuole. Another mutation could be at the pfmdr1 gene encoding for the transporter for importing solutes into the food vacuole, including the drugs mefloquine, halofantrine, and artemisinin (and possibly chloroquine), but this may not confer resistance on its own. In the laboratory, P. falciparum resistance to chloroquine can be reversed by combining it with various drugs such as calcium inhibitors, phenothiazines, antidepressants, and antihistamine compounds, but clinical evidence is limited and the usefulness of this approach in humans has not been established.[5]
| Drug | Mutations |
| Chloroquine | pfcrt (P. falciparum chloroquine resistance transporter, the key amino-acid change required to allow resistance to emerge has been identified as K76T), pfmdr1 (P. falciparum multidrug resistance transporter 1) |
| Quinine | pfcrt, pfmdr1, and pfnhe1 (P. falciparum sodium/proton exchanger 1) |
| Amodiaquine | pfcrt, pfmdr1 |
| Mefloquine | pfmdr1 |
| Piperaquine | pfcrt |
| Lumefantrine | pfcrt, pfmdr1 |
| Atovaquone | Single nucleotide polymorphisms in the cytochrome b gene |
| SP | Point mutations in pfdhfr and pfdhps |
| Artemisinin | Pf kelch protein gene on chromosome 13 (kelch13) |
| P. vivax | PvMDR1, pvdhfr and pvdhps |
Cross-resistance between the 4-aminoquinolines, chloroquine and amodiaquine, is common and development of resistance to mefloquine may also lead to resistance to halofantrine and quinine.[5]
Resistance to SP results from the mutations in the dihydro folate reductase (DHFR) and dihydropteroate synthase (DHPS) domains, respectively, with the parasites highly resistant to SP having a combination of triply mutated DHFR and doubly mutated DHPS. Resistance to atovaquone is related to point mutations in the cytochrome b gene, and emerges rapidly when it is used in monotherapy. Some cases of resistance to atovaquone and proguanil combination have also been reported, but all of these were not associated with mutation in the cytochrome b gene.[6]
Resistance to artemisinin derivatives has also been reported recently; a study published in July 2014 has reported that slowly clearing infections (clearance half-life >5 hours) were strongly associated with single point mutations in the “propeller” region of the P. falciparum kelch protein gene on chromosome 13 (kelch13).[7]
Origin and Distribution of antimalarial drug resistance
Drug-resistant strains have more often evolved out of areas of low malaria transmission, like Thailand, and then spread across to endemic areas like Africa, where they have contributed to worsening mortality.
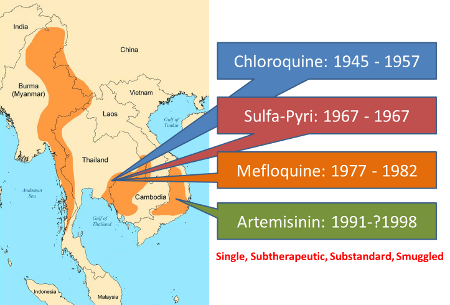
Origin of Drug Resistance in SE Asia
In 1955, WHO launched the most ambitious Global Malaria Eradication Programme; DDT was sprayed everywhere across Asia and South America and chloroquine was used extensively to treat malaria. In many areas, particularly in SE Asia and S America, mass administration of chloroquine was also started as a preventive measure and it was administered to even those who did not have malaria. As a result of this wide spread use, very soon chloroquine resistance in P. falciparum was observed; first in Thailand in 1957 and on the Colombia-Venezuela border in 1959. Pailin on Western Cambodia was a centre for gem mining in the 1950s having a lot of migrant labourers from Myanmar and Bangladesh. Chloroquine was used as mass prophylaxis to prevent malaria among these workers.[8] Through the returning workers, choloroquine resistance spread to Myanmar and Bangladesh 1969-70 and reached the neighbouring Karbi-Anglong district of Assam, India by 1973.[8-11] By 1980s, chloroquine resistance had spread to sub-Saharan Africa and today chloroquine has lost its efficacy in all but a few areas such as Central America (north-west of the Panama Canal), the island of Hispaniola, and limited areas of the Middle East and Central Asia.[5]
With chloroquine losing its efficacy, other drugs were introduced one after another in the SE Asia region, only to be lost to resistance in quick succession. Resistance to Sulfa-Pyrimethamine was reported in P. falciparum in the very year of its introduction in 1967, and it reached India by 1979 [4,9-11] and now it is frequently seen in South America and Africa.
During the Viet Nam War, the Chinese developed Artemisinin from the sweet worm wood; it turned out to be the fastest acting and safest antimalarial drug, and helped the north Vietnam army. During the same period, the American Army had developed mefloquine for its own soldiers, but it came to use after the war ended. Mefloquine was introduced in Thailand in 1977, but it became ineffective by 1982. Mefloquine resistance also meant cross-resistance to halofantrine and quinine.[5,12] Mefloquine resistance is now common in Southeast Asia and has also been reported from Amazon region of South America and Africa (sporadically).[5] Resistance to quinine has been reported from parts of Southeast Asia and South America.[5,13]
Viet Nam, poor by war, could not afford this expensive mefloquine of US and by 1991, developed its own production of artemisinin and started using it, largely as monotherapy, often in subtherapeutic doses. Later substandard artemesinin was smuggled to the entire region.
| Drug | Introduction | “First” year resistance reported | Difference (years) |
| Quinine | 1632 | 1910 | 278 |
| Chloroquine | 1945 | 1957 | 12 |
| Proguanil | 1948 | 1949 | 1 |
| Sulfadoxine-pyrimethamine | 1967 | 1967 | <1 |
| Mefloquine | 1977 | 1982 | 5 |
| Atovaquone | 1996 | 1996 | <1 |
| Artemisinins | 1971 | 1998? | 35 |
The Roll Back Malaria initiative was launched by WHO in 1998. By then artemisinin was gaining ground as the fastest acting antimalarial, and some early cases of recrudescence were being reported.[14] In view of this, WHO recommended a ban on Artemisinin monotherapy and in 2001, recommended Artemisinin Combination Therapy, combining a fast acting artemisinin compound with another slower acting antimalarial such as sulfa-pyrimethamine, mefloquine, amodiaquine or lumefantrine. The use of ACTs has since been going up all over the world, and as of now, this seems to be working, with 30% reduction in the incidence and about 47% reduction in mortality.[12] However, a study published in NEJM in July 2014 has reported increasing resistance in P. falciparum to ACTs in Thai-Cambodia border regions.[7] The study tracked 1241 children in 15 centres in 10 countries. The parasite clearance half lives were 1.9 h in Africa, but significantly prolonged at 7 hours in Thai-Cambodia border [2.3 hrs in Assam, India, 3.1 h in Myanmar, 3-6 hrs in Cambodia and 5-7 h in Thailand]. High proportion of patients in Thai-Cambodia had prolonged clearance and persisting parasitemia at 72 hours. The single point mutations in the “propeller” region of the P. falciparum kelch protein gene on chromosome 13 (kelch13), strongly associated with slowly clearing infections, were found to be widespread across Viet Nam, up to Myanmar. Thus, in this SE Asia region, short course ACT with artesunate are failing; however, ACTs at a longer 6 day course continue to be effective.[7]
Drug resistance in P. vivax malaria
Chloroquine resistance in P. vivax was first reported from Papua New Guinea in 1989 and since then, cases have been reported from Indonesia (Papua, Irian Jaya, and Island of Nias), Myanmar, India, Borneo, Guyana, parts of the Amazon Brazil, Columbia, Vietnam, Peru, Turkey, and Ethiopia. However, significant chloroquine resistance is confined largely to Indonesia, East Timor, Papua New Guinea, and other parts of Oceania, with highest prevalence (84%) of chloroquine resistant P. vivax reported from the Northeastern coast of Indonesian Papua. The World Malaria Report 2014 has indicated that P. vivax is resistant to chloroquine in at least 10 countries, but remains sensitive in most parts of Southeast Asia, the Indian subcontinent,the Korean peninsula, the Middle East, Northeast Africa, and most of South and Central America.[1,4,15,16]
Sulfa drugs are less active against P. vivax than against P. falciparum and there is widespread resistance of P. vivax to pyrimethamine. Therefore, the synergy of sulfa and pyrimethamine is not effective against P. vivax. The basis for the innate resistance of P. vivax to sulfa drugs is not clear, but it may be similar to the mechanism involved in P. falciparum. The wide use of sulfa drugs in the treatment of bacterial and plasmodial infections may have contributed to the selection of sulfa-resistant P. vivax populations.[17]
In recent years, there have been reports of failures of primaquine as antirelapse therapy for P. vivax malaria, from different regions including some parts of India. As the recurrence of symptoms and parasitemia following treatment of P. vivax infection could also be due to recrudescence of chloroquine-resistant strains or due to reinfection, it is difficult to confirm primaquine resistance in many of these cases. Some of the cases of treatment failure could also be due to incomplete or incorrect dosing or poor compliance. Although some studies have documented possible resistance to primaquine by polymerase chain reaction (PCR) single-strand conformational polymorphism, to differentiate relapses from reinfection, it is the resistance in tissue stages that is of concern rather than of the blood stages of the parasite.[18-20] However, in view of the reports of loss of efficacy of primaquine at 0.25 mg/kg/day for 14 days, there are suggestions to use primaquine at 0.5 mg/kg/day for 14 days instead.[18]
Drug resistance in India
In India, chloroquine-resistant P. falciparum malaria has been observed with increasing frequency across the country in recent years. Considering this, the Government of India has recommended the combination of artesunate and Sulfa-Pyrimethamine as the treatment of choice for P. falciparum (and mixed) infections all across the country.[21] In north east India, where resistance to SP has been documented, the National Drug Policy recommends the use of Artemether and Lumefantrine for the treatment of P. falciparum malaria. P. vivax remains sensitive to choloroquine all across the country.[21]
Identifying drug resistance
Estimation of treatment failure rate and validation of drug resistance can only be done by objective study of therapeutic efficacy. Such therapeutic efficacy studies are conducted in a controlled environment, in which drug administration is supervised, the results of microscopic examinations of blood films are validated, and the origin and quality of the drugs are verified.
The outcome of such a study is influenced by a combination of a human factor (immunity), a parasite factor (drug resistance) and individual variation leading to differences in the availability of the drug (pharmacokinetics). For example, an adult living in an area of high transmission might be able to eliminate resistant parasites even if the medicine is not fully effective, because of acquired immunity. Conversely, a non-immune child infected with drug-sensitive parasites who has severe gastrointestinal problems may experience therapeutic failure because of poor absorption. While therapeutic efficacy studies can help to predict the likelihood of drug resistance, additional tools are needed to confirm antimalarial drug resistance. First, it must be proven that the parasites are recrudescent in a patient who recently received treatment. The parasites are genotyped to distinguish between those that are recrudescent and those that caused a new infection. Evidence must be obtained that the patient had an adequate blood concentration of the drug or its metabolites, typically for at least four parasitic cycles. This can be confirmed by pharmacokinetic analyses of blood samples.[1]
There are different methods to identify and assess drug resistance in malaria. An in vivo assessment of drug sensitivity involves monitoring of the parasitological and/or clinical response following treatment. Case reports and passive detection of treatment failure may also be used to indicate drug resistance, but all cases of treatment failure may not be due to drug resistance; many factors such as incorrect dosing, noncompliance with duration of regimen, poor drug quality, drug interactions, poor or erratic absorption, and misdiagnosis can all contribute to treatment failure.[5]
A standardized in vivo test protocol for assessing the response of P. falciparum to chloroquine was first developed in 1965 and after many revisions, the latest protocol has been put forth by the WHO in 2003.[22] It involves a fairly simple, prospective evaluation of the clinical and parasitological response to treatment for uncomplicated malaria. It also provides for measuring blood levels of the drugs, extending the period of follow-up, and testing for molecular markers to help distinguish reinfection from recrudescence whenever technically and logistically feasible. Worsening or less than expected resolution of clinical features and parasite density by third day after treatment is considered as early treatment failure. Worsening or reappearance of symptoms after day 4 and persistence of parasitemia at day 28 (day 14 in intense transmission areas) are considered as late treatment failure.[22]
 In areas with intense transmission, recurrence of symptoms after day 14 may be due to reinfection and molecular tools such as PCR may be needed to differentiate it from recrudescence due to treatment failure. As younger children often have a less favorable therapeutic response to antimalarial drugs than do older children and adults, the evaluation of antimalarials for uncomplicated malaria should emphasize treatment efficacy in children <5 years with clinically apparent malaria.[22] Wherever possible, blood or plasma levels of the antimalarial should also be measured in prospective assessments, so that drug resistance can be distinguished from treatment failures due to pharmacokinetic reasons.[1,22]
In areas with intense transmission, recurrence of symptoms after day 14 may be due to reinfection and molecular tools such as PCR may be needed to differentiate it from recrudescence due to treatment failure. As younger children often have a less favorable therapeutic response to antimalarial drugs than do older children and adults, the evaluation of antimalarials for uncomplicated malaria should emphasize treatment efficacy in children <5 years with clinically apparent malaria.[22] Wherever possible, blood or plasma levels of the antimalarial should also be measured in prospective assessments, so that drug resistance can be distinguished from treatment failures due to pharmacokinetic reasons.[1,22]
Drug resistance can also be studied by in vitro methods, done by exposing finger-prick blood samples on microtitre plates to precisely known quantities of drug and observing for inhibition of maturation into schizonts. Animal model studies with in vivo tests conducted in nonhuman animal models and molecular tests using PCR to indicate the presence of mutations encoding drug resistance are the other methods used.[1] The results of these additional tests must, however, be interpreted with caution, as they do not always correlate well with the results of therapeutic efficacy studies, and the predictive usefulness of some of these tests remains to be defined.[1]
References
- Guidelines for the Treatment of Malaria. 2nd edn. WHO, Geneva, 2010. Available at http://whqlibdoc.who.int/publications/2010/9789241547925_eng.pdf
- Maguire JD, Sumawinata IW, Masbar S, et al. Chloroquine-resistant Plasmodium malariae in south Sumatra, Indonesia. Lancet. 2002;360(9326):58–60. doi:10.1016/S0140-6736(02)09336-4.
- Young MD. Resistance of Plasmodium malariae to pyrimethamine (daraprim). Am J Trop Med Hyg. 1957;6(4):621–624.
- WHO. Global report on antimalarial drug efficacy and drug resistance: 2000-2010. WHO. Geneva. 2010. At http://whqlibdoc.who.int/publications/2010/9789241500470_eng.pdf
- Bloland PB. Drug resistance in malaria. WHO/CDS/CSR/DRS/2001.4. World Health Organization. Geneva. 2001. Available at http://www.who.int/csr/resources/publications/drugresist/malaria.pdf
- Hyde JE. Drug-resistant malaria – an insight. FEBS J. 2007;274:4688–4698. doi:10.1111/j.1742-4658.2007.05999.x
- Ashley EA, Dhorda M, Fairhurst RM et al. Spread of Artemisinin Resistance in Plasmodium falciparum Malaria. N Engl J Med July 2014; 371:411-423. At http://www.nejm.org/doi/full/10.1056/NEJMoa1314981
- Packard RM. The Origins of Antimalarial-Drug Resistance. N Engl J Med. 2014;371:397-399. At http://www.nejm.org/doi/full/10.1056/NEJMp1403340
- Farooq U, Mahajan RC. Drugresistance in malaria. J Vector Borne Dis. 2004 Sep-Dec;41(3-4):45-53. At http://www.mrcindia.org/journal/issues/413045.pdf
- Shah NK, Dhillon GPS, Dash AP et al. Antimalarial drug resistance of P. falciparum in India: changes over time and space. Lancet Infect Dis. Jan 2011; 11(1): 57-64. At http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3068018/
- Sehgal PN, Sharma MID, Sharma SL, Gogai S. Resistance to chloroquine in falciparum malaria in Assam state, India. J Commun Dis 1973; 5:175-80
- World Health Organization. Antimicrobial resistance: global report on surveillance. 2014. Athttp://apps.who.int/iris/bitstream/10665/112642/1/9789241564748_eng.pdf
- Wongsrichanalai C, Pickard AL, Wernsdorfer WH, Meshnick SR. Epidemiology of drug-resistant malaria. The Lancet Infectious Diseases. 2002;2(4):209-218. At http://www.thelancet.com/journals/laninf/article/PIIS1473-3099(02)00239-6/fulltext
- Luxemburger C, AB, Silamut K, Nosten F, van Vugt M, Gimenez F, Chongsuphajaisiddhi T, White NJ. Two patients with falciparum malaria and poor in vivo responses to artesunate. Trans R Soc Trop Med Hyg 1998;92: 668–669. Full Text
- Teka H, Petros B, Yamuah L, et al. Chloroquine-resistant Plasmodium vivax malaria in Debre Zeit, Ethiopia. Malaria J. 2008;7:220. doi:10.1186/1475-2875-7-220.
- Baird JK. Chloroquine resistance in Plasmodium vivax. Antimicrob Agents Chemother. 2004;48(11):4075–4083. doi: 10.1128/AAC.48.11.4075-4083.2004.
- Korsinczky M, Fischer K, Chen N, et al. Sulfadoxine resistance in Plasmodium vivax is associated with a specific amino acid in dihydropteroate synthase at the putative sulfadoxine-binding site. Antimicrob Agents Chemother. 2004;48(6):2214–2222. doi:10.1128/AAC.48.6.2214–2222.2004.
- Kshirsagar NA. Malaria: Antimalarial resistance and policy ramifications and challenges. J Postgrad Med. 2006;52(4):291–293.
- Baird JK, Hoffman SL. Primaquine therapy for malaria. Clin Infect Dis. 2004;39:1336–1345.
- Gogtay NJ, Kshirsagar NA, Vaidya AB. Current challenges in drug-resistant malaria. J Postgrad Med. 2006;52:241–242
- National Drug Policy on Malaria – 2013. Directorate of National Vector Borne Disease Control Programme. Govt. of India. New Delhi. 2013. Available at http://nvbdcp.gov.in/Doc/National-Drug-Policy-2013.pdf
- Assessment and Monitoring of Antimalarial Drug Efficacy for the Treatment of Uncomplicated Falciparum Malaria. WHO/HTM/RBM/2003.50. World Health Organization. Geneva. 2003.
©malariasite.com ©BS Kakkilaya | Last Updated: Mar 23, 2015